paired end sequencing insert size
PE reads generally have a smaller insert size 1kp than MP 2-5 kb. Note that the paired distance you set does not need to be exact for all reads.

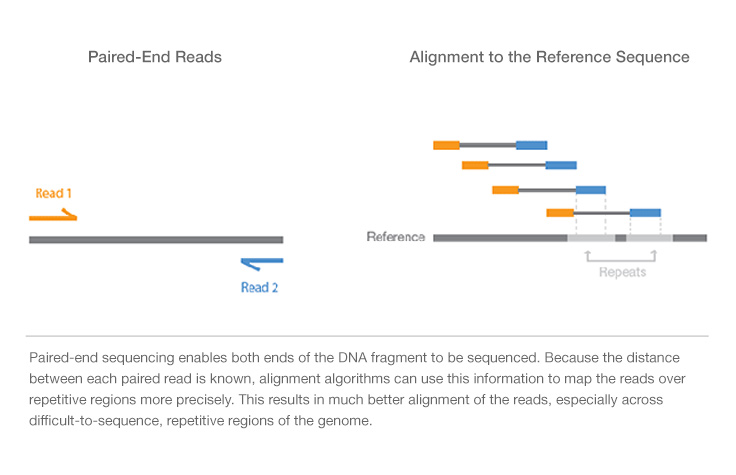
Interpreting Color By Insert Size Integrative Genomics Viewer
The maximum distance x for a pair considered to be properly paired SAM flag 02 is calculated by solving equation Phix-musigmaxLp0 where mu is the mean sigma is the standard error of the insert size distribution L is the length of the genome p0 is prior of anomalous pair and Phi is the standard cumulative distribution function.
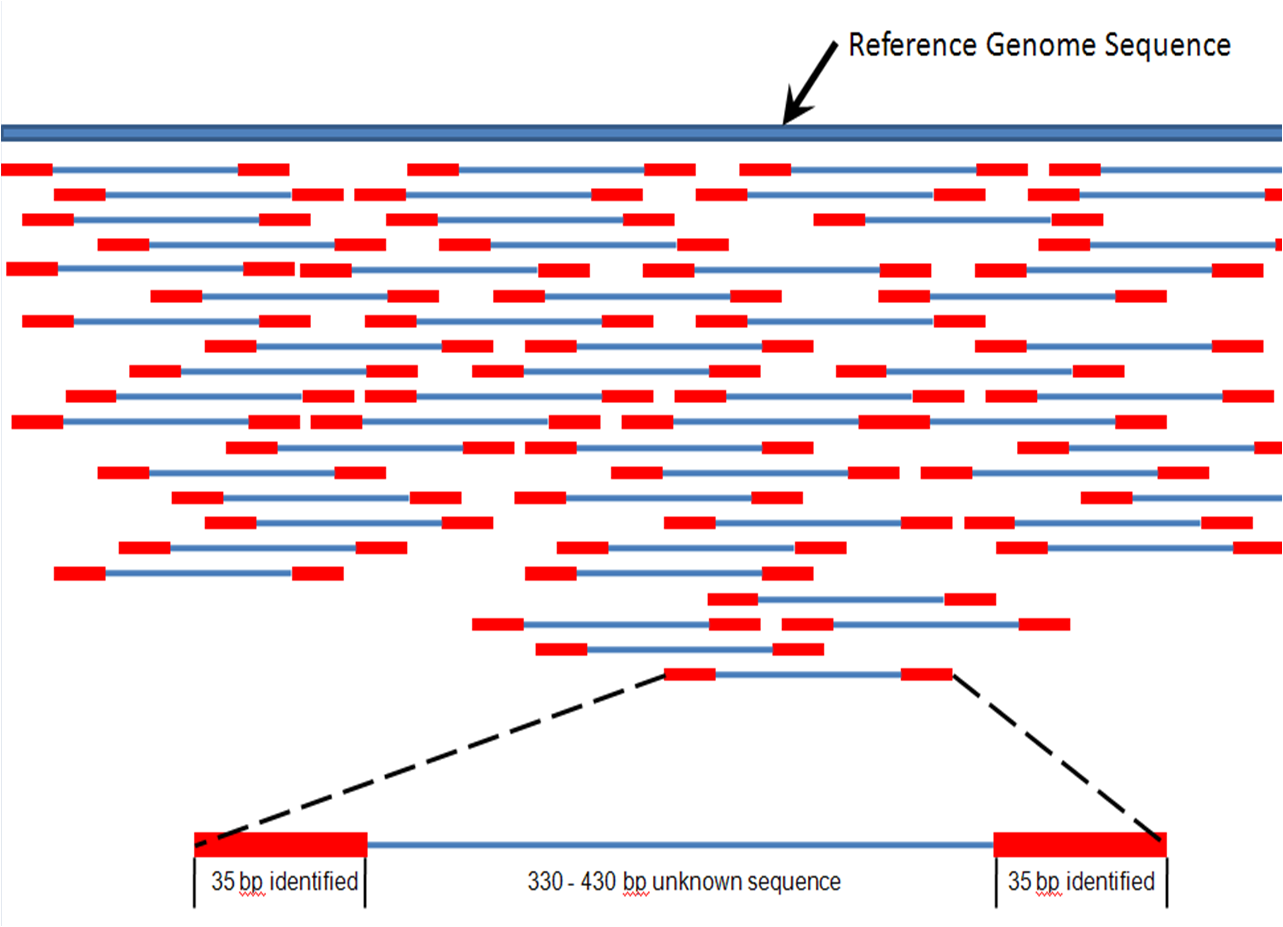
. Including flanking intronic regions of diagnostic significance eg due to splice variants of about 30bp at either side the average target of interest is about 220 bp long. Bioc-sig-seq Calculating insert size for paired-end mate pair samples Pratap Abhishek APratap at somumarylandedu Thu Jan 21 181908 CET 2010. The products of this ligation reaction are purified and sizeselected by agarose gel electrophoresis.
2Align paired-end RNA-Seq data to the reference transcripts using minimum insert-length as zero and maximum insert-length as 500 or more. Can be used for. For Illumina systems DNA insert size range of 200800 bp.
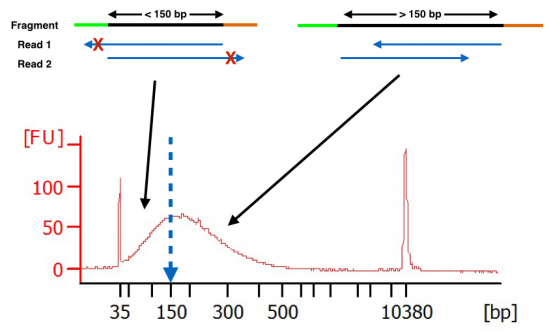
If processing a small file set the minimum percentage option M to 05 otherwise an error may occur. So why you get reads of the same length when. Normally the insert size is longer than the sum of the two read lengths meaning there is an unsequenced inner part in the middle of the insert.
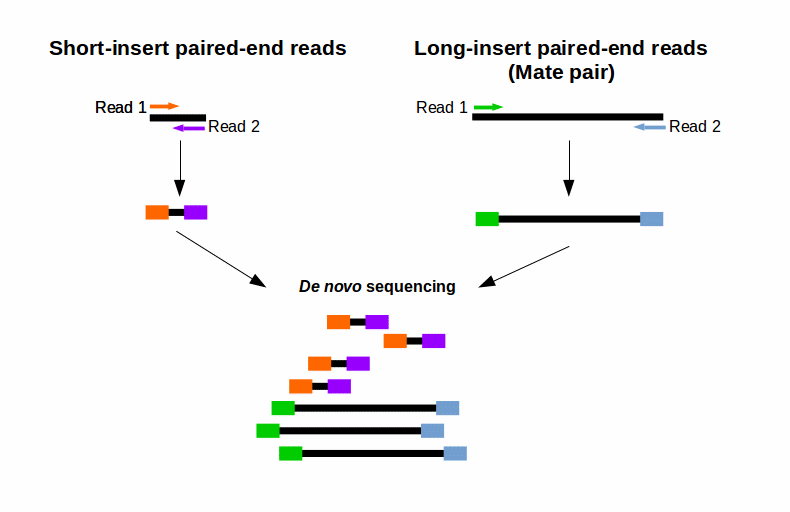
I wrote a single Python script to estimate the paired-end read insert length or fragment length from read mapping information ie SAMBAM files. What is the optimal insert size. Paired-end sequencing also provides greater ability to overcome the obstacles of characterizing repetitive sequence ele-ments by filling in gaps of consensus sequence to achieve complete.
Please see InsertSizeMetrics for detailed explanations of each metric. Does not require methylation of DNA or restriction digestion. However the average size of human coding exons is only 160bp.
-I 0 -X 500 BWA. Despite the benefits of gaining SS-PE data with paired ends of varying distance the standard Illumina protocol allows only non-strand-specific paired-end sequencing with a single insert size. Paired ends of varying distance the standard Illumina protocol allows only non-strand-specific paired-end sequencing with a single insert size.
Set output format as SAMBowTie or BAMBWA. Standard paired-end libraries 200500 bp can be used to detect large and small insertions deletions indels inversions and other rearrangements. Since paired-end sequencing cannot resolve repeats longer than the insert size bridges which attempt to span long repeats cannot be trusted.
Broad Range of Applications. Requires the same amount of DNA as single-read genomic DNA or cDNA sequencing. Simple workflow allows generation of unique ranges of insert sizes.
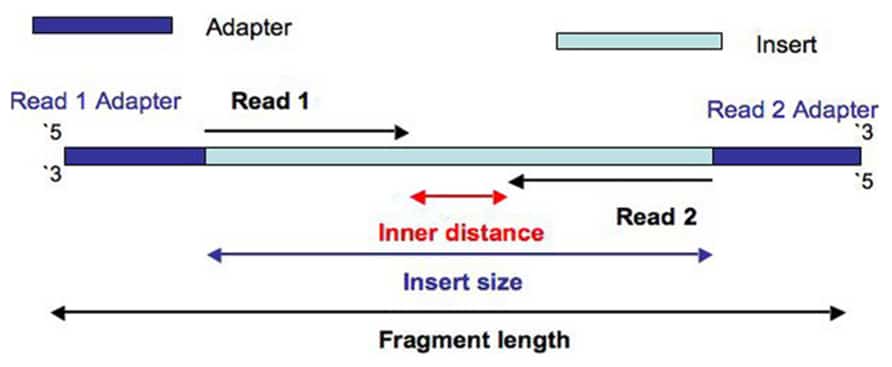
The difference in insert size stems from the difference in protocols. For instance for a PE150 sequencing run the insert size should be above 300bp. The sequencing starts at Read 1 Adapter mate 1 and ends with the sequencing from Read 2 Adapter mate 2.
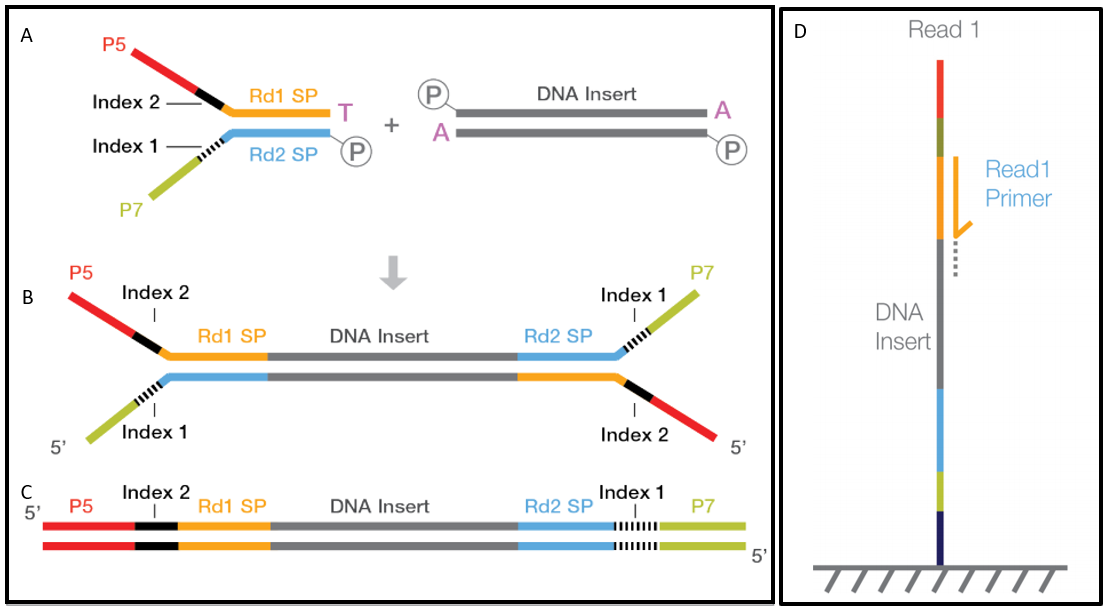
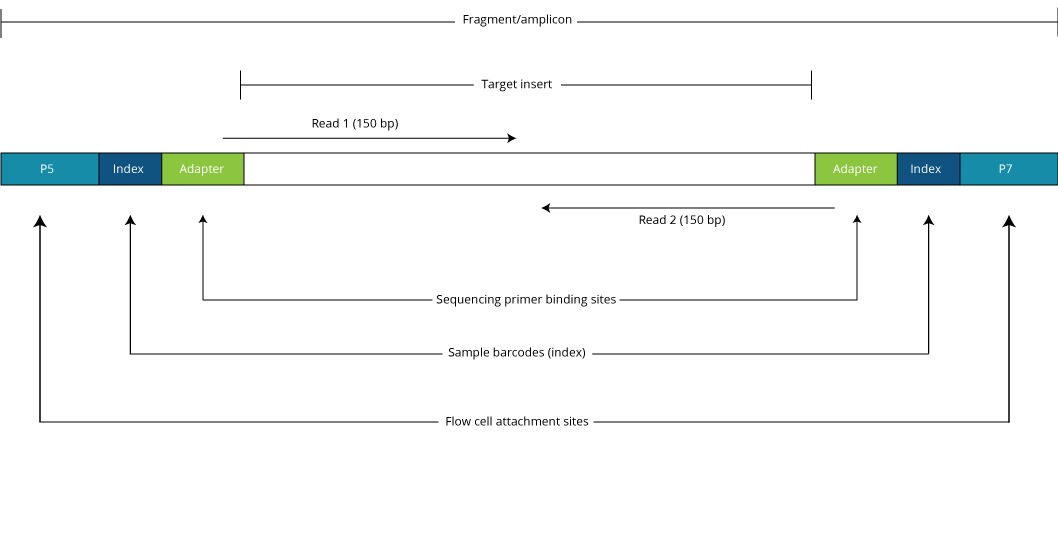
The DNA between adapter the sequences is the insert. Geneious will still assemble reads that are under or over the expected distance but the pairing information will be used to help the assembler resolve complex placement issues. Despite the benefits of gaining SS-PE data with paired ends of varying distance the standard Illumina protocol allows only non-strand-specific paired-end sequencing with a single insert size.
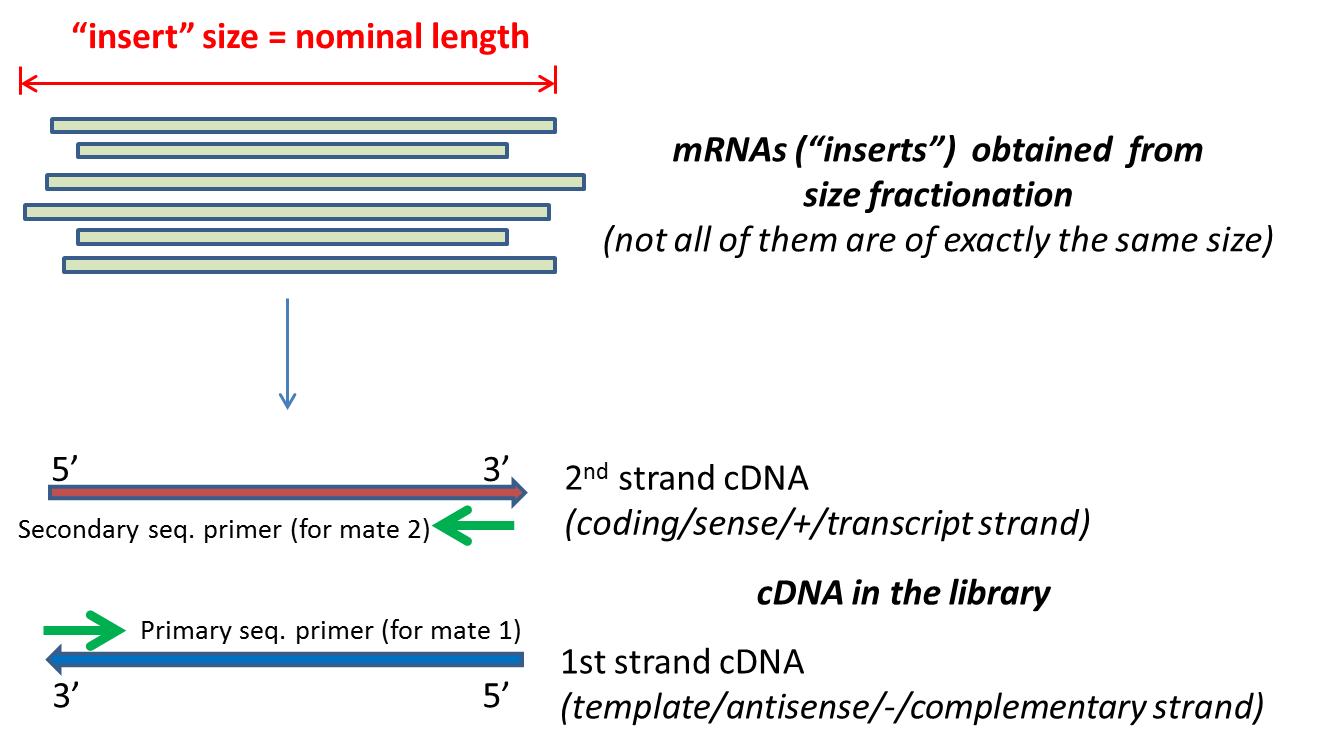
These reads can then be sequenced using the same SP1-SP2 adapter protocols used in PE sequencing. If you have a range of insert sizes you should set it on the mid point of the range. Paired end sequencing refers to the fact that the fragment s sequenced were sequenced from both ends and not just the one as was true for first.
The variation of insert sizes is often large and the average size difficult to control. The algorithm is simple. Typically Illumina paired end reads have an insert longer than the combined length of both reads see Figure Figure1 1.
Insert size -- The insert size refers to the distance between the pairs. Here we modify the Illumina RNA ligation protocol to allow SS-PE sequencing by using a custom pre-adenylated 3 adaptor. Bioc-sig-seq Calculating insert size for paired-end mate pair samples.
Check the TLEN field in the SAM format throw out pair-end reads whose pairs are too far away and use them to estimate the mean and variance of the insert length. Ad Gene Expression Profiling Chromosome Counting Epigenetic Changes Molecular Analysis. Here we modify the Illumina RNA ligation protocol to allow SS-PE sequencing by using a custom pre-adenylated 3 adaptor.
Fast and Accurate Next-Generation Sequencing Results Enabled by Ion Torrent Technology. To read more about the different parts of a prepared DNA fragment please take a look on the figure in the article about observed sequence lengths in Illumina. The length of this sequence is known as the insert size not to be.
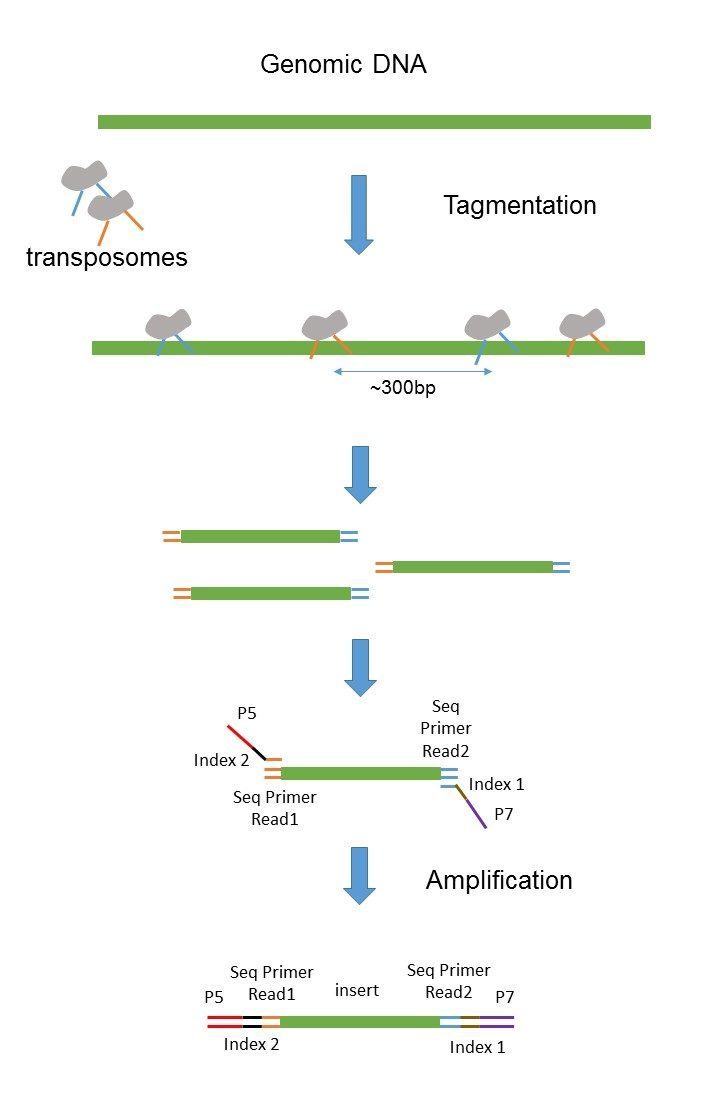
The insert is normally the stretch of sequence between the paired-end adapters so in your case the insert size would be 250 bp 2x75 bp reads 100 bp unsequenced middle piece. The Illumina NexteraXT transposon protocol is a cost effective way to generate paired end libraries. The fragment size which you need to select for during a gel purification for example would be the insert size length of both adapters around 120 bp extra for both Illumina.
It depends on the used Illumina system the used reagents kits and the mode singlepaired end. This can result in a proportion of fragments with an insert size of less than the length of a single read. Using the latest Illumina platform the MiSeq paired end reads of.
Java -jar picardjar CollectInsertSizeMetrics. Transpososomes are used to fragment DNA to be sequenced and add adapter sequences in a single step known as tagmentation. Figure 1 Sequencing Library after PairedEnd Sample Preparation The two adapters contain sequences that are complimentary to the two surfacebound amplification primers on the flow cells.
The reads have a length of typically 50 - 300 bp. Bridges are given a quality score most importantly based on the length of the bridge compared to the length of the paired end insert size so bridges which span a long repeat are given a low score. We generate parallel libraries with differing.

Five Approaches To Detect Cnvs From Ngs Short Reads A Paired End Download Scientific Diagram
What Is Mate Pair Sequencing For

Diagram To Show The Construction Of A Fragment With An Insert Size Is Download Scientific Diagram

The Insert Size In Paired End Data Seqanswers

Detecting Deletion Events A When Mapping Paired End Reads To The Download Scientific Diagram

Paired End Illumina Reads
What Are Paired End Reads The Sequencing Center
Annotare Embl Ebi
Ngs
What Read Lengths Are Produced By Modern Illumina Sequencers

Diagram To Show The Construction Of A Fragment With An Insert Size Is Download Scientific Diagram

Definition Of Essential Terms A Definition Of Fragment Read And Download Scientific Diagram

Interpreting Color By Insert Size Integrative Genomics Viewer

Library Insert Size Libraries Were Constructed Using The Standard A Download Scientific Diagram
What Happens When Hardware Advances Faster Than Molecular Protocols Cofactor Genomics

Pdf Assessment Of Insert Sizes And Adapter Content In Fastq Data From Nexteraxt Libraries
Paired End Sequencing France Genomique
How Short Inserts Affect Sequencing Performance
What Is Paired End 150 Omega Bioservices